Болезни альфа и бета
Талассемия — болезнь, вызванная гемоглобином
Талассемия — это не одна, а группа похожих болезней, передающихся по наследству и вызванных нарушенным синтезом белковой части гемоглобина. Заболевание достается ребенку с вероятностью в 100%, если один из родителей содержит измененный ген. Поэтому оно считается самой распространенной наследственной патологией.
Наибольшее число случаев зарегистрировано в странах, расположенных по берегам Средиземного моря, на африканском континенте, в Средней Азии, Индии. Среди населения Азербайджана каждый десятый имеет измененные гены.
Немного истории
Болезнь была впервые описана в 1925 году американскими врачами-педиатрами, которые при лечении эмигрантов из Италии выявили одинаковую клиническую картину у детей с тяжелым малокровием, увеличением печени и селезенки, изменениями костей.
Затем появились работы, описывающие более легкое течение болезни у взрослых пациентов. Термин «талассемия» предложили в 1936 году. Дословно он означает «болезнь морского побережья». Высказана мысль о связи патогенеза заболевания с нарушением синтеза глобиновых цепочек.
Почему разрушаются эритроциты?
Его структура состоит из пигмента (гема), включающего железо, и набора из двух пар белковых цепочек. Их называют по типичному расположению аминокислот альфа- и бета-цепями. При нарушенном синтезе одного из типов полипептидов накапливается другой вид.
В результате разрушаются все клетки эритроцитарного ряда (сами эритроциты и их предшественники), из них выходит гемоглобин. Малокровие (анемия) развивается по гипохромному типу. Это подтверждается низким цветовым показателем.
«Виновниками» разрушений являются гены, ответственные за построение белковой части гемоглобина. Их мутация нарушает способность составлять необходимый набор аминокислот в цепи. Причину изменений связывают с возбудителем малярии — плазмодием. Доказаны его мутирующие способности. Талассемия по территории распространенности совпадает с эпидемическими зонами малярии.
Дети могут получить болезнь путем наследования от родителей в двух видах:
- гомозиготном — ген-мутант передается от обоих родителей;
- гетерозиготном — ген болезни передается только от матери или отца, носителем может быть один из родителей.
Соответственно, называются формы талассемии.
При гетерозиготном носительстве выделяют:
Разновидности заболевания
Все виды заболевания связаны с кислородной недостаточностью, наступившей вследствие разрушения эритроцитов — единственных клеток, обеспечивающих газообмен в органах и тканях.
В зависимости от сбоя синтеза полипептидной цепочки различают:
- альфа-талассемию;
- бета-талассемию.
Наиболее распространена бета-талассемия. Она характеризуется избыточным накоплением α-цепочек глобина.
Выявлены также редкие формы болезни, которые названы гамма- и дельта-талассемией. В группу включены:
- некоторые гемоглобинопатии;
- гомозиготная альфа-талассемия с водянкой плода;
- смешанный вид β- и δ-талассемии, когда поражаются одновременно дельта- и бета-цепи.
Каждая форма имеет свои типичные клинические проявления и отличия. Но в зависимости от тяжести течения принято выделять:
- хроническую легкую — люди доживают до старости;
- хроническую среднетяжелую — пациенты не переживают детский возраст;
- тяжелую — ребенок погибает в период новорожденности.
Клинические проявления большой талассемии
Симптомы талассемии зависят по клинике и времени проявления от вида генной мутации.
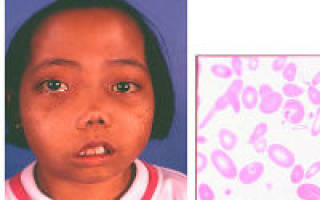
Большая талассемия (болезнь Кули) развивается при гомозиготной передаче от родителей. Ее проявления заметны у детей сразу после рождения.
У новорожденного замечают:
- удлиненный вверх череп («башенный»);
- более развитую верхнюю челюсть;
- монголоидный тип лицевого скелета.
К 12-месячному возрасту проявляется:
- расширенная носовая перегородка, «приплюснутый» нос;
- костные наросты на стопах;
- нарушенный прикус;
- желтушность кожи (в связи с поражением селезенки).
Кислородное голодание тканей приводит к органным нарушениям. При осмотре отмечаются:
- увеличение печени, край плотный на ощупь (развивается ранний цирроз);
- возникновение сердечной недостаточности (лишнее железо откладывается в миокарде и поражает сократимость сердечной мышцы);
- отставание в физическом и умственном развитии от сверстников.
У малыша развивается сахарный диабет из-за фиброзирования поджелудочной железы и печеночная недостаточность. В тяжелых случаях он живет не более одного года.
В старшем возрасте проявляются такие осложнения:
- трофические язвы на коже, вызванные локальными нарушениями кровообращения;
- частые переломы костей;
- повторные воспаления в легких;
- калькулезный холецистит;
- кардиосклероз;
- сепсис при контакте с инфекцией;
- нарушенное полового развитие.
Клиника малой талассемии
Малая или гетерозиготная форма передается одним из родителей. Второй «здоровый» ген сглаживает повреждения. Симптомы талассемии могут длительно не проявляться или вызывать признаки, общие с другими заболеваниями:
- слабость, повышенную утомляемость;
- головные боли;
- головокружение.
При осмотре обращается внимание на следующее:
- бледность кожи с желтушным оттенком;
- возможно увеличение печени и селезенки.
У ребенка с малой формой резко увеличена восприимчивость к инфекциям, снижен иммунитет. Очень тяжело и часто протекают вирусные и кишечные инфекционные заболевания.
Во взрослом состоянии у женщины при беременности развивается водянка плода (повышение внутричерепного давления и скопление жидкости в желудочках мозга). Эта патология несовместима с жизнью ребенка, в случае нормальных родов ведет к тяжелым неврологическим и психическим отклонениям.
Минимальной β-талассемией называют синдром Сильвестрони — Бьянко. Эта форма заболевания протекает практически бессимптомно, обнаруживается случайно в семьях со случаями талассемии.
Диагностика
Диагностика талассемии должна начинаться с генетической консультации супругов перед зачатием ребенка. Во время беременности при необходимости проводят анализ амниотической жидкости. Уже на ранних сроках в эритроцитах плода можно обнаружить характерные изменения.
Порой внешний осмотр и выяснение семейного анамнеза позволяют заподозрить болезнь у ребенка без лабораторной диагностики.
В анализе крови выявляют:
- снижение уровня гемоглобина до 30–50 г/л при гомозиготном варианте и до 90 –110 г/л при гетерозиготном;
- низкий цветовой показатель (менее 0,5), который образуется малым насыщением клеток гемоглобином);
- рост ретикулоцитов (предшественников эритроцитов) до 4%.
При просмотре окрашенного мазка под микроскопом обращают внимание на следующее:
- наличие слабо окрашенных (гипохромных) эритроцитов;
- изменение размеров и формы красных клеток, эритроциты приобретают формы овала, серпа, шара.
Одна из характерных разновидностей серповидных клеток — дрепаноциты.
Биохимические тесты показывают нарушенный обмен железа, как при гемолитической анемии:
- высокий уровень свободного билирубина;
- повышенную концентрацию железа в сыворотке;
- сниженную способность к связыванию железа.
Исследование особенностей гемоглобина проводят с помощью ацетат-целлюлозной пленки. Подобный анализ позволяет определить количественный уровень фракций. Гомозиготная бета-талассемия отличается увеличенным уровнем фетального гемоглобина (в норме он содержится у плода, а у взрослого человека всего до 1%), неспособного переносить кислород.
Использование метода полимеразной цепной реакции позволяет изучить строение полипептидных цепочек гемоглобина.
Генетический анализ выявляет мутацию в одиннадцатой паре хромосом при β-талассемии, другие специфические изменения, типичные для прочих форм.
Исследование пунктата костного мозга проводится для выявления повышенного содержания незрелых эритроцитов в виде сидеробластов.
Рентгенологическое исследование способствует обнаружению дефектов костной ткани:
- участков остеопороза (сниженной плотности);
- увеличенной массы костей черепа;
- поперечной исчерченности на мелких костях кистей и стоп;
- на рентгенограмме черепа при большой форме β-талассемии видно типичное игольчатое поражение надкостницы, которое называется симптомом «волосатого черепа».
Ультразвуковое исследование подтверждает увеличение печени и селезенки, помогает обнаружить камни в желчевыводящей системе.
Лабораторные данные более отчетливо выражены при β-талассемии. Другие формы болезни не дают четкой картины.
Заболевания, с которыми необходимо проводить дифференциальную диагностику:
- железодефицитная анемия,
- серповидно-клеточная анемия,
- гемолитическая аутоиммунная анемия,
- наследственный микросфероцитоз.
Генетическая интерпретация видов бета-талассемии
Ученые-генетики выяснили интересную закономерность: люди имеют одинаковую мутацию генов, отвечающих за синтез гемоглобина, но клиника и степень выраженности заболевания у них отличаются.
Гены могут находиться в следующих состояниях:
- нормальные — характерны для здорового человека;
- поврежденные частично — «работает» неполно, из-за чего синтез полипептидных цепей недостаточен;
- разрушенные полностью — синтез останавливается.
По этому признаку виды талассемии делят на:
- минор — самая легкая форма, поврежден только один ген, внешне человек выглядит здоровым, по анализу крови можно предположить небольшую анемию;
- интермедиа — недостаток бета-цепей серьезно сказывается на синтезе, эритроциты недоразвиты, малокровие выражено с явными признаками, но организм еще может приспособиться, поэтому отсутствует необходимость в постоянных переливаниях крови;
- майор — мутации подверглись все гены, больному необходимы постоянные переливания крови по жизненным показаниям.
Генетические разновидности альфа-талассемии
По охвату мутацией генов, их локусов (определенных частей, отрезков), отвечающих за синтез альфа-цепочек полипептидов, предлагается выделение нескольких групп заболевания:
- мутация только в одном локусе — клинических проявлений нет;
- изменения двух (пары) локусов в одном или разных генах — анализ крови показывает снижение гемоглобина, уменьшение размера эритроцитов;
- поражение трех локусов — выражается в кислородной гипоксии органов, увеличении селезенки, возникновении Н гемоглобинопатии, образующийся гемоглобин нестоек, разлагаясь, вызывает гемолитическую анемию;
- мутация всех локусов — полностью прекращает синтез альфа-цепей, при подобной ситуации происходит внутриутробная гибель плода или ребенок умирает сразу после родов.
Генетические исследования подтвердили также неодинаковое значение пар генов, одна пара из двух является главной, а другая имеет второстепенную роль. Клинические проявления зависят от того, в какой паре произошла мутация.
Проблемы терапии
Лечение талассемии зависит от тяжести поражения эритропоэза, степени охвата генов процессами мутации. В настоящее время применяются следующие методы.
Диетический рацион направлен на снижение всасывания железа в кишечнике, рекомендуются орехи, какао, соя, чай.
Терапия дополняется ежедневным устранением излишков железа с помощью введения (хелатов — специальных комплексов, повышающих действие лекарства). Для связывания железа назначается Десферал. Этот препарат предупреждает сидероз (патологическое состояние, вызванное отложением в тканях железа), но на уровень гемоглобина не влияет.
При возникновении резкого ухудшения состояния по аналогии с гемолитическими кризами показаны глюкокортикоиды в больших дозах.
Спленэктомия возможна при больших размерах селезенки детям после пятилетнего возраста. Наиболее оптимален возраст 8–10 лет. После удаления селезенки наступает период улучшения, но опасен риск присоединения инфекции.
Для трансплантации необходим донор, совпадающий по всем параметрам, лучше всего из близких родственников.
К симптоматическим средствам относятся препараты гепатопротекторного действия, большие дозы аскорбинки помогают вывести излишки железа из организма.
Все формы талассемии нуждаются в препаратах с фолиевой кислотой и витаминами группы В. На фоне присоединившейся инфекции, при беременности следует применять фолиевую кислоту в больших дозах, поскольку неэффективное кроветворения при талассемии значительно увеличивает ее потребление клетками.
Можно ли предупредить заболевание?
Талассемия считается до настоящего времени неизлечимым заболеванием. К мерам предупреждения относят дородовую диагностику для решения вопроса о наследовании признаков, своевременного прерывания беременности.
Рожденный ребенок с легкой степенью болезни может прожить всю жизнь без тяжелых последствий. Но гарантировать именно эту форму патологии современная наука не может. Особенно следует подумать о целесообразности деторождения парам с гомозиготной наследственностью.
Методика исследования клеток плода во время беременности (забор осуществляется путем прокола матки через брюшную стенку) опасна возможным занесением инфекции и преждевременными родами. Поэтому лучше решать проблемы в медико-генетической консультации до плановой беременности.
Больных с талассемией ведут и наблюдают совместно гематологи, педиатры и терапевты.
Бета-талассемия
Талассемия — группа тяжелых генетических заболеваний крови, при которых нарушается синтез жизненно важного белка гемоглобина, входящего в состав эритроцитов, обеспечивающих кислородом все ткани и органы. Болезнь развивается из-за нарушения синтеза гемоглобина, связанного со сниженной продукцией одной из полипептидных цепей глобина (альфа, бета,гамма, дельта). Если нарушается синтез альфа-глобиновых цепей, болезнь классифицируют как альфа-талассемию. В случае недостаточного синтеза бета-глобиновых пептидных цепей, патологию определяют как бета-талассемию.
Заболеваемость альфа-талассемией распространена в южно-азиатских и западноафриканских странах. Бета-талассемия чаще встречается в Средиземноморских регионах, а также станах Северной Африки западной Азии. Ежегодно по всему миру рождается более 280 тысяч детей с синдромом талассемии.
Если вам необходима консультация специалиста по орфанным заболеваниям, заполнив все поля формы, и наши консультанты будут рады Вам помочь.
Что такое бета-талассемия
По данным ВОЗ, бета-талассемия является наиболее распространенной формой из группы наследственных заболеваний крови по аутосомно-рецессивному типу. В равной степени затрагивает как женщин, так и мужчин. Болезнь дифференцируют на гомозиготную форму, когда генетически она наследуется от одного из родителей и гетерозиготную форму, в случае наследования мутантного гена от обоих родителей.
Основная причина возникновения заболевания — мутации гена, который отвечает за синтез полипептидной цепи гемоглобина. Дефект на молекулярном уровне обусловлен неэффективным процессом или мутацией регуляторных генов, соединением аномальной матричной РНК или хромосомные перестройки структурных генов.
Механизм развития бета-талассемии заключается в недостаточном количестве синтезируемых бета-цепей, что приводит к избыточному продуцированию альфа-цепей, которые откладываются в клетках эритроидного ряда и повреждают их. Процесс сопровождается гибелью ретикулоцитов в селезенке, разрушением эритроцитов и деструкцией эритробластов в костном мозге, а также накоплением фетального гемоглобина. В результате этого сокращается транспортировка кислорода по всем тканям и органам, возникает хроническая анемия и тканевая гипоксия, что в свою очередь становится причиной многих заболеваний, в том числе костных деформаций и т.д.
МКБ-10
В класс МКБ-10 входят заболевания крови и кроветворных органов, а также гемолитические анемии, талассемии и тяжелая бета-талассемия (анемия Кули). По международной классификации болезней из группы МКБ-10, бета-талассемии присвоен код D56.1.
Симптомы
Клиническая картина при бета-талассемии проявляется у детей в течение 1-2 лет после рождения. Гомозиготная форма болезни сопровождается слабостью, хронической усталостью, повышением температуры тела, одышкой, бледностью и желтушностью кожного покрова, интоксикацией. Вследствие хронической анемии у детей наблюдается значительная задержка в физическом и умственном развитии. Нередко при бета-талассемии обнаруживаются нарушения в работе сердца, печени, эндокринных желез, что вызвано патологическим накоплением железа в организме. Из-за экстрамедуллярного кроветворения и тотального разрушения эритроцитов увеличивается селезенка и печень.
На фоне поражения трубчатых костей возникают патологические переломы, развивается синовит крупных суставов, желчнокаменная болезнь. Пациенты с бета-талассемией подвержены частым инфекционным болезням. Гомозиготная форма может привести к серьезным осложнениям: кардиосклерозу, сахарному диабету, сердечной недостаточности, циррозу печени, сепсису, пневмонии.
Бета-талассемия гетерозиготной формы может протекать бессимптомно или с незначительными проявлениями: повышенной утомляемостью на фоне гипохромной анемии, головокружениями, небольшим увеличением селезенки, головными болями.
Признаки
Уже в первый год после рождения, у детей с большой бета-талассемией можно отметить внешние признаки заболевания. Это монголоидный овал лица, характерный разрез глаз, четырехугольная (башенная) форма черепа, расширенная носовая перегородка (седловидная переносица), увеличенная верхняя челюсть, неправильный прикус. Наблюдается изменения скелетной структуры, иногда могут образовываться изъязвления в области голеней на фоне желтушности кожи. У маленьких детей достаточно выраженные внешние признаки бета-талассемии, что дает основания для предварительного диагноза.
Диагностика
При подозрениях на бета-талассемию специалисты в первую очередь изучают семейный анамнез, учитывают жалобы пациента и проводят физикальный осмотр. Для постановки диагноза требуется комплексное обследование, состоящее из таких процедур:
- Лабораторные исследования крови, позволяющие определить уровень железа, концентрацию билирубина, средний объем эритроцитов, нарушения цепей гемоглобина и другие показатели.
- Молекулярно-генетические исследования на предмет генных мутаций.
- Пункция костного мозга, подтверждающая гиперплазию красного кроветворного ростка.
- Краниография черепа, выявляющая игольчатый периостоз (симптом волосатого черепа).
- УЗИ брюшной полости — для оценки состояния желчевыводящих путей, а также размеров печени и селезенки.
В процессе обследования бета-талассемию дифференцируют от аутоиммунной, железодефицитной и серповидно-клеточной анемии, а также от наследственного микросфероцитоза. Немаловажное значение имеет пренатальная диагностика. Женщинам, у которых в семье есть родственники с подобным диагнозом, необходимо провести забор амниотической жидкости, чтобы подтвердить или опровергнуть наличие генетической патологии у плода. Данное исследование позволит начать своевременное лечение.
Классификация
В зависимости от генетических факторов и степени тяжести симптоматической картины, бета-талассемию разделяют на 3 группы:
Малая бета-талассемия — легкая форма заболевания, когда полной или частичной мутации подвержен только 1 ген, отвечающий за синтез бета-цепей. В анамнезе у носителей этой формы болезни отмечается лишь небольшой уровень эритроцитов в крови и легкая степень анемии.
Большая бета-талассемия — тяжелая форма патологии, при которой мутация затрагивает оба гена, ответственных за синтез полипептидных цепей гемоглобина. При большой бета-талассемии у детей до года, существует высокий риск летального исхода.
Промежуточная бета-талассемия — отличается большим дефицитом бета-цепей, может переходить в тяжелую форму болезни. В большинстве случаев протекает без серьезных нарушений в организме с минимально выраженной клинической картиной.
Лечение бета-талассемии
Тактика лечения зависит от формы и степени тяжести талассемии. Пациенты с малой бета-талассемией в лечении не нуждаются и могут самостоятельно компенсировать последствия врожденного заболевания, соблюдая здоровый образ жизни (отказ от вредных привычек, двигательная активность, сбалансированное питание).
При большой и промежуточной бета-талассемии врачи применяют следующие методы лечения:
- Трансфузионная терапия — переливание донорской крови. В случае промежуточной формы бета-талассемии трансфузию проводят редко, при сердечной недостаточности, легочной гипертензии, тяжелых костных деформациях, значительной задержке роста. Большая форма заболевания требует регулярного переливания эритроцитов (1-2 раза в месяц).
- Хелаторная терапия — введение препаратов, связывающих железо и выводящих его из организма. При гемолитических кризах используют глюкокортикоиды.
- Спленэктомия — удаление селезенки в случае ее гипертрофии.
- Пересадка костного мозга (ТКМ) — трансплантация донорских стволовых клеток, способствует возобновлению нормальной работы кроветворной системы.
Также пациентам со всеми формами бета-талассемии показан прием витаминов В, С, Е и фолиевой кислоты. Для предотвращения инфекционных осложнений требуется вакцинация.
Прогноз заболевания и профилактика
После пересадки костного мозга многие пациенты полностью выздоравливают. Постоянное переливание эритроцитной массы увеличивает продолжительность жизни маленьким детям, у которых была выявлена большая бета-талассемия. При гетерозиготной форме заболевания с бессимптомным течением качество и продолжительность жизни не страдают.
Профилактика бета-талассемии заключается в планировании беременности будущих родителей. Супружеской паре необходимо обязательно проконсультироваться с генетиком, если у кого-то из родственников была выявлена анемия Кули. При высоком генетическом риске носители мутантных генов должны отказаться от деторождения. Также к первичной профилактике бета-талассемии относят дородовую диагностику плода.
Если вам необходима консультация специалиста по орфанным заболеваниям, заполнив все поля формы, и наши консультанты будут рады Вам помочь.
Талассемия – виды, описание, причины заболевания
Понятие «талассемия» объединяет ряд генетических заболеваний крови. Чтобы понять, что представляет собой талассемия, необходимо понять, как устроена кровь. Гемоглобин является компонентом, переносящим кислород в эритроциты. Он состоит из двух различных белков — альфа и бета. Если организм человека не производит достаточное количество какого-либо из этих белков, красные кровяные клетки не формируются должным образом и не могут переносить достаточное количество кислорода. В результате у человека развивается анемия, которая может начаться в раннем возрасте и продолжается в течение всей жизни.
Талассемия — это группа родственных расстройств, которые имеют ряд общих и ряд отличительных признаков. Различия между типами талассемии обусловливают те симптомы, которые испытывает человек при развитии этого заболевания.
Из-за преимущественного распространения заболевания среди средиземноморского контингента, талассемию, а точнее различные её виды, называют средиземноморской анемией.
Альфа-талассемия
Люди, чей гемоглобин не производит в достаточном количестве альфа-белок, страдают от альфа-талассемии.
Существует несколько типов альфа-талассемии:
1. Бессимптомный.
Это состояние, как правило, не вызывает никаких проблем со здоровьем, поскольку уровень отсутствия альфа-белка очень мал. Функция гемоглобина при этом не нарушается. Человек, больной таким заболеванием, называется «молчаливым носителем». У таких людей могут родиться дети с признаками альфа-талассемии.
2. Мутация альфа-гемоглобина.
Наблюдается, в основном, на территории Ямайки, где впервые было обнаружено это состояние крови. Проблем со здоровьем у таких пациентов обычно нет.
3. Мягкая форма альфа-талассемии.
В этом состоянии уровень отсутствия альфа-белка несколько выше. Пациенты с такими симптомами имеют умеренную форму альфа-талассемии, ошибочно принимаемую многими врачами за железодефицитную анемию.
4. Синдром Н-гемоглобина.
В этом состоянии у пациентов наблюдаются серьезные проблемы со здоровьем, например, увеличение селезенки, вирусные инфекции. Эта форма заболевания названа в честь аномального гемоглобина Н, который разрушает красные кровяные клетки.
5. Хронический Н-гемоглобин.
Более опасный для здоровья человека синдром, приводящий к появлению серьезной анемии, проблемам с внутренними органами.
6. Гомозиготный Н-гемоглобин.
Этот симптом наблюдается у детей, чьи родители являются носителями хронической формы Н-гемоглобина.
Еще одна форма альфа-талассемии называется водянкой плода. При этом состоянии у новорожденного отсутствуют в ДНК альфа-гены, которые трансформируют гамма-глобины, произведенные организмом, в гемоглобин Барта.
Большинство детей с таким состоянием умирают до или сразу после рождения. В некоторых, крайне редких случаях, внутриутробное переливание крови позволяют ребенку с водянкой родиться живым, однако таким детям требуется пожизненное переливание крови и медицинская помощь.
Бета-талассемия
Люди, чей гемоглобин не производит достаточное количество бета-белка, болеют бета-талассемией. Заболевание чаще встречается у людей средиземноморского происхождения, например, итальянцев, греков, а также обитателей Аравийского полуострова, Ирана, Африки, Юго-Восточной Азии и южной части Китая.
Существует три типа бета-талассемии, которые варьируются от легкой до тяжелой степени, в зависимости от их воздействия на организм.
Типы бета-талассемии
1. Малая бета-талассемия.
Отсутствие бета-белка недостаточно велико, чтобы вызвать проблемы с нормальной функцией гемоглобина. Человек с этим состоянием просто является генетическим носителем признаков талассемии, и с точки зрения врачей практически здоров. Возможна умеренная степень железодефицитной анемии.
Как и при мягкой форме альфа-талассемии, врачи часто ошибочно принимают небольшие эритроциты с бета-талассемией за проявление железодефицитной анемии и назначают препараты железа.
2. Промежуточная талассемия.
Достаточно большой уровень отсутствия бета-белка в гемоглобине, что вызывает анемию умеренной степени, деформацию костей, увеличение селезенки.
3. Бета-талассемия третьей степени (анемия Кули).
Самая тяжелая форма бета-талассемии, при которой в гемоглобине полностью отсутствует бета-белок. Такая форма заболевания является условием для регулярных переливаний крови. Такие обширные переливания крови приводят к перенасыщению железом. Это является условием для хелатотерапии, целью которой является предотвращение ранней смерти от полиорганной недостаточности.
Другие формы талассемии
1. Дельта-бета-талассемия — еще одна форма бета-талассемии, характеризующаяся полным отсутствием или понижением уровня синтеза дельта и бета-цепей глобина с компенсаторным увеличением экспрессии синтеза гамма-цепей. Распространенность этой формы заболевания неизвестна, однако чаще всего она встречается в Греции и Италии.
Гетерозиготная форма заболевания протекает клинически бессимптомно, с мягким микроцитозом и повышением уровня гемоглобина HbA2. При гетерозиготной наследственной бета-талассемии у больных производится только гамма-глобин с образованием HbF.
2. E-бета-талассемия.
Гемоглобин Е является одним из наиболее распространенных аномальных гемоглобинов. Заболевание проявляется у жителей Юго-Восточной Азии, например, камбоджийцев, вьетнамцев и тайцев. В сочетании с бета-талассемией, гемоглобин Е вызывает умеренно-тяжелую анемию, которая схожа с симптомами промежуточной бета-талассемии.
3. Серповидная-бета-талассемия.
Обусловлена сочетанием бета-талассемии и гемоглобина S, аномального гемоглобина, свойственного людям, больным серповидно-клеточной анемией. Это заболевание характерно для греков, турков, итальянцев. Чем больше бета-глобина производит бета-ген, тем тяжелее заболевание и его симптомы.
Симптомы талассемии
Талассемия в слабой степени обычно не вызывает никаких симптомов. Если всё же симптомы и есть, они напоминают мягкую форму анемии.
Наиболее выражены симптомы анемии Кули. Они проявляются на второй год жизни ребенка. Кроме проявлений железодефицитной анемии, возможны такие симптомы:
- частые вирусные инфекции;
- бледность;
- плохой аппетит;
- желтуха;
- увеличение внутренних органов.
Гемоглобинопатия
Гемоглобин производится генами, которые контролируют экспрессию этого белка. Дефекты в этих генах могут производить аномальные типы гемоглобина и приводят к анемии, которую называют гемоглобинопатия. Аномальные типы белка-гемоглобина появляются в таких случаях:
1. Структурные дефекты в молекуле гемоглобина.
Случается так, что мутации изменяют одну из аминокислот. Чаще всего такое изменение безобидно и не нарушает ни структуру, ни функцию молекул гемоглобина. Однако случается и так, что изменение одной аминокислоты резко нарушает поведение молекулы гемоглобина и приводит к болезням.
2. Уменьшение производства одной из двух субъединиц молекул гемоглобина.
Равные количества гемоглобина альфа- и бета-цепей являются необходимыми для нормальной функции крови. Дисбаланс в цепи гемоглобина повреждает и разрушает эритроциты, тем самым приводя к анемии.
3. Аномальные соединения нормальных субъединиц.
Одна субъединица альфа-цепи и одна субъединица бета-цепи объединяются, чтобы произвести нормальный гемоглобин. В тяжелых случаях субъединицы глобина начинают объединяться в группы по четверо. Они неактивны функционально и не транспортируют кислород. При этом альфа-субъединицы быстро деградируют при отсутствии партнера типа бета.
Наследственное персистирование фетального гемоглобина (НПФГ)
Персистенция (персистирование) фетального гемоглобина (HPFH, НПФГ) является состоянием, при котором производство фетального гемоглобина (гемоглобина F), продолжается и в зрелом возрасте.
Фетальный гемоглобин — это основной вид гемоглобина, который вырабатывается у плода во время нахождения в утробе матери. Наличие такого элемента в организме позволяет при достаточно малом количестве крови в организме плода выполнять кислородоснабжающую функцию.
Когда ребенок рождается, гемоглобин-Ф заменяется гемоглобином-А, его более «взрослой» формой. Если же замена происходит не полностью, это свидетельствует от талассемии.
Причины
НПФГ вызвана мутациями в генах β-глобина. Процент таких мутаций обычно равен 10-15%. В основном это состояние бессимптомно и обнаруживается во время скрининга, причиной которого является другое расстройство крови.
По материалам:
1.Pr Renzo GALANELLO, Dr Raffaella ORIGA
2.Gretchen Holm, George Krucik, MD
3.The Cooley’s Anemia Foundation
4.National Institutes of Health, Department of Health and Human Services
5.wikipedia.org
Почему продукты с трансжирами в составе нас медленно убивают?
Почему некоторые песни заедают у нас в голове на весь день?
Как не отравиться домашней пищей?
Как не заразиться коронавирусом во время путешествия?
Что делать, если вы подозреваете у себя коронавирус?
Как запах, которого мы не слышим, влияет на выбор сексуального партнёра
Как правильно хранить лекарства
Дефицит серотонина – это опасно
Папилломы (бородавки) на интимных местах
Что такое пренатальный скрининг и зачем его проводят
А, альфа и бета вашего здоровья и красоты
В нашей стране первый витамин, которые люди сами себе чаще всего назначают – витамин А. Его пьют для цикла, кожи, при выпадении волос, и просто для профилактики пока неведомых болячек.
Некоторые «особо выдающиеся» врачи назначают ретинол даже женщинам при подготовке к беременности…И мало кто знает, что чистый аптечный витамин А – довольно токсичный ретинол, имеющий серьезные побочки при передозировке и нужно знать когда, сколько и для чего можно пить без риска.
Ретинол (истинный витамин A) — жирорастворимый витамин, который совершенно не синтезируется в организме и должен ежедневно восполняться с едой. В продуктах питания он довольно нестабилен и разрушается при термообработке (до 35%) и в значительной степени при длительном хранении продуктов.
Главные пищевые источники – печень трески (консервы) и рыбий жир, говяжья и куриная печень, сливочное масло (дорогое, не из пальмового масла), сыр, сметана, и намного меньше его в молоке, яичном желтке. Но чтобы вы понимали сколько нужно съесть этих продуктов для покрытия дефицита скажу, что дневная норма вит А это 1300 мл сметаны или банка консервированной печени трески. Выходит добрать его из питания, и при этом не ожиреть, маловероятно.
Симптомы дефицита витамина А
Самый точный признак дефицита вит. А — куриная слепота, хотя никталопия звучит, конечно, благороднее. Протестировать себя на ее наличие вы можете уже сегодня ночью — достаточно просто выключить свет…Долго не получается сориентировать в пространстве, а при включении света перед глазами появляются цветные пятна?? Товарищ, тебе не хватает витамина А!
Никталопия не так и страшна для большинства из нас, но опасна для жизни, если вы за рулем – чуть стемнеет и риск попасть в аварию у вас повышен. Но проблема решается назначением ударных доз витамин А и B2 (рибофлавин, продается в каждой аптеке) на 5-6 недель.
Кстати, в биокоррекции аутизма есть интересный протокол «мегадоз витамина А», при котором всего пару дней ребенку даются ультравысокие дозы ретинола. Многие отмечают, что после этого дети становятся более контактными (начинают смотреть в глаза). Объяснение этому простое: ретинол входит в состав чувствительных к свету рецепторов в сетчатке глаза. При дефиците витамина А смотреть в глаза собеседнику или на свет физически просто некомфортно даже взрослому человеку.
Второй признак дефицита витамина А – глобальная сухость: волос, кожи, слизистых, шелушение и растрескивание кожи, синдром сухого глаза, иногда и расслоение ногтей. Но этот признак ненадежный – практически те же симптомы характерны для железодефицитной анемии и гипотиреоза.
Интересен другой момент: тироксин, принимаемый при гипотиреозе – антагонист вит. А, то есть впоследствии приводит к его дефициту. На фоне его приема (и при гипертиреозе) нужно регулярно восполнять вит А. Уверена, эндокринолог забыл вас предупредить об этом.
Третья группа симптомов – задержка роста, плохой аппетит, частые простуды и ячмени (!!) на глазах у детей и взрослых. Хотя тоже симптомы не специфические.
Дефициту витамина А способствуют:
- диета с сокращением или исключением жиров (возможно «сидите» вы на ней ненамеренно, просто ваши привычные продукты не содержат достаточное количество жиров)
- дефицит цинка (вот почему часто появляется и акне, цинк и А в этом случае принимают вместе в очень больших дозировках)
- болезни кишечника (особенно болезнь Крона), желудка (Хеликобактер в том числе), желчного, печени (нарушается усвоение витамина) – некоторые язвенные и воспалительные заболевания жкт лечат даже раствором ретинола – пример тому аптечный «Аекол»
- прием тироксина – выше уже писала, что параллельно нужен прием вит. А
- дефицит витамина Е и белка, а так же белковые диеты
- периоды повышенной потребности в витамине А – беременность, кормление, смена климата, тяжелая физическая работа, либо активные занятия фитнесом, период активного роста у детей.
- алкоголь, причем неважно что вы пьете – водку стаканами или французское винцо по вечерам – выводится витамин А с одинаковой скоростью, и даже один из способов снять интоксикацию витамином А – выпить любой алкогольный напиток.
Как восполнить дефицит витамина А?
Есть два пути – через питание и дополнительный прием. Если симптомы уже проявились или у вас есть болезни жкт, то питанием вы не вылечитесь – 5-6 недельные курсы витаминных препаратов несколько раз в год обязательны. И дозировки в этом случае могут превышать в 20 раз дневную норму. Питание – лишь профилактика гиповитаминоза.
На витаминных препаратах всегда приводятся дозировки витаминов и % от рекомендованной нормы потребления. В США норма — 5000 ME(EU) или 1500 мкг вит. А в сутки, в России – 900 мкг, но 1400 для кормящих мам и 1000 для детей.. Норма – это минимум, который позволяет не уйти витаминам в минус в вашем организме и спровоцировать гипо- и авитаминоз. В России при этом нормы ниже западных, да и там чаще расчет ведется уже от 10000 ME(EU) или 3000 мкг.
Но вот лечебные дозировки иные – в зависимости от проблемы, они варьируются от 50 000 до 300 000 ME. Поэтому не стоит удивляться тому, что на некоторых БАД написано, к примеру, 600% от нормы или все 1000% — значит, велик шанс получить даже лечебный эффект.
Рабочая дозировка при лечении акне от 100 000 до 300 000 МЕ сроком до 12-16 недель – некое подобие Роаккутана (одна из форм витамина А), но с меньшими побочками и без последствий в дальнейшем. Хотя, спешить с беременностью после такого лечения не стоит, да и выпадение волос в конце курса может начаться.
Выпадение и ретинол – это отдельная тема. Волосы практически никогда не выпадают из-за дефицита витамина А , зато его длительный прием в больших дозировках может спровоцировать диффузный падеж. Следовательно, при выпадении лучше всего выбирать витаминные комплексы, дозировки в которых не далеки от дневных норм, либо ретинол заменен на безопасный бета-каротин.
При себорее (жирной, сухой, себорейном дерматите) ретинол может значительно улучшить ситуацию, но не затягивайте лечебный курс более чем на 2 месяца.
Витамин А выпускается в основном в двух формах – ретинола ацетат и более редкий ретинола пальмитат. Ацетат усваивается хуже и ему в организме надо метаболизироваться в пальмитат. Очень важно принимать ретинол либо на ночь, либо рано утром – так он намного быстрее достигнет органов – мишеней.
Ретинол пальмитат есть далеко не в каждой аптеке.
На айхерб (все названия бад кликабельны)
У приема витамина А в ретиноловой форме есть свои противопоказания:
- курение…печаль совсем с курящими –вит. А выводит их организм стремительнее, но его дополнительный прием у них же еще больше повышает риск развития рака…придется выбирать – или сигаретка/или витаминка
- вит А в форме аптечного ретинола не рекомендуется принимать при подготовке к беременности – он тератогенен, а после длительного курса лучше даже пару месяцев не думать о беременности
- онкология — витамины ускоряют метаболические процессы, и в итоге в организме все растет и развивается лучше, в том числе и уже имеющиеся опухоли
- прием ОК с ретинолом так же не очень стыкуется из-за способности стероидных гормонов искусственно повышать в плазме уровень витамина А, именно девушки, сидящие на ОК чаще всего получают ретиноловые интоксикации.
С ретинолом разобрались – принимать его можно, но соблюдая осторожность. Переходим к провитаминам витамина А, из них организм синтезирует витамин А самостоятельно в нужных ему количествах и передозировки невозможны.
Провитамины группы А объединены в группу каротиноидов – мощных антиоксидантов, в число которых в ходят бета- и альфа-каротин, гамма-каротин, лютеин, ликопин, астаксантин и зеаксантин. Напомню, что антиоксиданты обезвреживают свободные радикалы, приводящие к повреждению клеток, последующему старению и развитию тяжелых заболеваний. Считается, что сдерживая деятельность свободных радикалов, мы отодвигаем старость, морщины и онкологию.
Несмотря на то, что витамина А (ретинол) относится к антиоксидантам, по ряду причин он на деле им не является и для омоложения кожи и организма всегда лучше каротиноиды, чем чистый ретинол.
Только альфа — и бета – каротин из этой каротиновой компашки преобразуются в значительной степени в витамин A. Они депонируются в печени и метаболизируется в витамин А по мере надобности, не приводя к интоксикации. Лично я советую выбирать витаминные комплексы, в которых витамин А представлен минимум на 50 % бета-каротином, а для беременных – и все 100.
Дозировки бета-каротина так же делятся на восполняющие и лечебные (выше нормы в несколько раз). Всем известный эффект пожелтения кожи после переедания морковки, который чем-то схож с загаром, происходит как раз из-за переизбытка запаса каротиноидов в печени и не представляет вреда для здоровья. Есть даже БАД для загара без солнца, работающие по этому принципу.
На айхерб ничего подобного я не нашла, но много их выпускается во Франции: Forté Pharma Expert SelfTan, Arkopharma Phytobronz Sun-Preparing и пр. Самый большой выбор на cocooncenter.co.uk.
Ученые уже давно доказано, что и альфа и бета-каротин предотвращают заболевания сердца и сосудов, защищают от рака (в частности, от рака шейки матки), не дают развиться катаракте, различным кожным заболеваниям и борются с возрастными явлениями (старение и сухость кожи) и т.д.. В общем, эффект еще внушительнее, чем от приема витамина А, но без последствий.
Где искать альфа- и бета-каротин
В темно-зеленых, оранжевых и желтых фруктах и овощах, особенно в моркови, тыкве, дыне, брокколи, абрикосах, капусте, шпинате, манго, киви, морских водорослях и, конечно, же хлорелле.
Натуральные каротиноиды гораздо предпочтительнее синтетических, и если не получается ежедневно употреблять овощи и фрукты в достаточном количестве, то отдавайте предпочтение витаминам, полученным из природного сырья – моркови, брокколи, и д.р..Часто на упаковке таких добавок пишут -1 таблетка равна такому-то количеству овощей.
Интересная особенность: вареные морковь и шпинат содержат каротиноидов в несколько раз больше, чем сырые. Так же помните, что бета- и альфа -каротин лучше всего усваиваются в сочетании с витамином Е, цинком и селеном. Жиры для усвоения тоже нужны – по этой причине в тертую морковь и добавляют сметану.
Кому необходим альфа — и бета — каротин
В первую очередь всем страдающим болезнями желчного пузыря, печени и поджелудочной, болезнью Крона, принимающим препараты для понижения уровня холестерина, лечения псориаза и артрита. Так же людям с пониженным иммунитетом, снижающимся зрением, любыми кожными заболеваниями и выпадением волос – каротиноиды его никогда не усугубляют.
Что попить и где купить
Веторон (бета-каротин, вит Е и С)– прекрасный российский препарат, доступный в каждой аптеке нашей страны.
Масло льняное «Морковное» и «Масло Клеопатры» (с добавлением морковного) производитель «Компас Здоровья», ищите в интернет – магазинах и Лавках Здоровья
БАД из серии «Живые витамины» от Родника здоровья ( Морковь с кабачком)
Что за болезнь талассемия?
Часто дети могут унаследовать от своих родителей не только характерные черты и положительные качества, но также и наследственное заболевание.
Вероятность получения какой-либо наследственной болезни зависит напрямую от наличия неполноценных генов в родителях. Талассемия относится именно к таким генетическим заболеваниям, которые передаются по наследству от родителей к их детям.
Что такое талассемия
Талассемия – это группа генетических заболеваний крови (код по МКБ 10 – D56), вызывающая неправильную мутацию эритроцитов, вследствие чего происходит нарушение транспортировки молекул кислорода к клеткам и тканям организма.
Гемоглобин составляет основную массу кровяного тельца и является главным веществом для связывания молекул кислорода и их перемещения по всему организму. При этом заболевании нарушается весь процесс синтеза полипептидных цепей гемоглобина, что влечет к неполноценной и короткой работоспособности эритроцитов.
В результате таких изменений происходит нехватка кислорода в тканях организма, что приводит к патологии и ухудшению функциональности органов человека.
Формы талассемии
За работоспособность гемоглобина отвечают полипептидные цепи: альфа (α) и бета (β). Нарушение синтеза в любой из этих цепей может привести к возникновению аномальных эритроцитов.
Различают два типа этого заболевания по неправильной аномалии полипептидных цепей:
- альфа-талассемия (код по МКБ 10 – D56.0);
- бета-талассемия (код по МКБ 10 – D56.1).
А также любой вид талассемии может отличаться по способу приобретения гена-мутанта от родителей. Гетерозиготная талассемия приобретается только от одного родителя и обычно носит легкую форму заболевания. Гомозиготная талассемия приобретается сразу от обоих родителей и может носить среднюю или тяжелую форму заболевания.
Альфа талассемия
За построение альфа-цепи отвечают четыре гена (по два гена от каждого родителя).
От количества переданных «больных» генов будет зависеть степень тяжести заболевания талассемии:
- Мутация в одном гене. Заболевание не обнаруживается в связи с отсутствием клинических проявлений. Человек с такой формой талассемии может быть только носителем, что в будущем приведет к вероятной передаче заболевания своим детям.
- Мутация в двух генах. Малая форма талассемии характеризуется низким уровнем гемоглобина в составе крови и небольшими размерами эритроцитов. Может приобретаться от одного или обоих родителей.
- Мутация в трех генах. Средняя форма заболевания, имеет явное нарушение кислородного питания организма. Могут возникать различные патологии в системах и органах человеческого организма.
- Мутация в четырех генах. Большая форма заболевания или альфа-талассемия майор. При такой аномалии полностью отсутствуют альфа-цепи, отвечающие за перенос молекул кислорода в клетки. Такая форма заболевания приводит к гибели ребенка еще в утробе матери или в первые дни после рождения.
Бета талассемия
В создании бета-цепи в гемоглобине участвуют только два гена (по одному от каждого родителя). При возникновении мутации одного или двух генов этой цепи появляется бета-талассемия.
Различают три формы этого вида заболевания:
- Минор или малая форма талассемии возникает при наличии одно гена-мутанта. Характеризуется малозаметным снижением бета-цепей, что влияет на отсутствие внешних признаков заболевания.
При анализах крови обнаруживаются малые размеры красных кровяных телец. Человек с такой формой заболевания проживает относительно здоровую жизнь. - Интермедиа или средняя форма талассемии образуется при незначительной мутации двух генов. Проявляются значительные повреждения бета-цепей, что сказывается на очень низком уровне гемоглобина и наличия большого количества недоразвитых эритроцитов. Без отсутствия лечения средняя форма обычно переходит в тяжелую.
- Майор или большая форма талассемии (анемия Кули) появляется при полной мутации обоих генов, отвечающих за бета-цепи. При этой форме требуется постоянное переливание крови для постоянного поддержания уровня гемоглобина.
Симптомы
При малой и большой формах талассемии симптомы проявляются по-разному. Малая талассемия по способу приобретения относится к гетерозиготной, то есть ген талассемии передается от одного родителя. В таком случае из-за здорового гена от второго родителя вся общая картина заболевания будет незначительно заметна.
К основным симптомам малой формы можно отнести:
- общая усталость, вялость и слабость;
- частые головные боли и головокружения;
- бледность и желтизна кожного покрова;
- скопление газов, метеоризм;
- высокая восприимчивость к вирусным и кишечным инфекциям;
- возможно увеличение селезенки и печени при пальпации и УЗИ диагностике.
Средние и тяжелые формы талассемии относятся к гомозиготным, так как неполноценные гены получают от отца и матери сразу.
Симптомы при таких формах заболевания являются ярко выраженными:
- башневидная форма черепа;
- образование монголоидных черт лица;
- увеличение верхней челюсти и расширение носовой перегородки на протяжении протекания болезни;
- желтизна и бледность кожи. Также читайте нашу статью про бледность лица при беременности.
- наблюдаются умственное и физическое отставание в развитии;
- образование костных наростов на стопах;
- увеличение селезенки и печени из-за нехватки кислорода;
Дети при тяжелой талассемии редко переступают пятилетний рубеж.
У больных при переходе в подростковый возраст появляются дополнительные симптомы:
- частые переломы костей;
- частые инфекции легких;
- сепсис при контакте с инфекцией
- отсталое половое развитие.
Диагностика
Первым шагом является своевременная генетическая консультация перед планированием беременности для выяснения наследственности талассемии у будущих родителей. Пренатальная диагностика возможна уже на одиннадцатой неделе беременности. Ранее обнаружение заболевания поможет заблаговременно начать бороться с талассемией.
Иногда история семейного анамнеза и внешний осмотр пациента позволяет заподозрить болезнь крови на ранних сроках.
Для подтверждения диагноза и начала лечения назначают следующие анализы при талассемии:
- общий анализ крови – проверяется уровень гемоглобина, размеры и формы эритроцитов, наличие ретикулоцитов.
- биохимия крови – проверка уровня билирубина, концентрация железа, способность к связыванию железа.
- ПЦР – выявление неполноценного гена талассемии в хромосоме. Позволяет окончательно подтвердить или отвергнуть диагноз талассемия.
- УЗИ-диагностика – проводится осмотр размеров печени, селезенки и наличие билирубиновых камней.
- пункция костного мозга – состав и характер сбоя системы кровообразования.
- рентгенологическое исследование – обнаружение дефектов скелета и костной ткани.
- специфические анализы крови – определение повреждений системы кровообразования.
Причины заболевания
- Талассемия является самым сложным наследственным заболеванием, так как передается от родителей к детям через неполноценные гены, подвергшиеся мутации. Иногда родители, являясь носителями, могут не подозревать о существовании этого заболевания в своей крови. Но талассемия в полной мере может проявить себя уже в переданных детям генах.
- При наличии неправильных генов в хромосомах нарушается синтез гемоглобина, что приводит к потере функциональности эритроцитов.
- Очаги распространения этого заболевания приходятся в основном на Средиземноморье, страны Ближнего Востока и в средней Африке, крайне редко в Латинской Америке.
- Возникновение первостепенной мутации гена родителя происходит из-за малярийного плазмодия. Научно доказано, что именно плазмодий при попадании в кровь человека оказывает основное влияние на мутацию генов в шестнадцатой и одиннадцатой парах хромосом, отвечающих за создание альфа- и бета-цепей.
Лечение
Талассемия до сих пор относится к ряду редко излечимых заболеваний. Обусловлено это в первую очередь слабой эффективностью и сложностью методики лечения.
При малой форме талассемии не требуется особое лечение, но существует план мероприятий по поддержанию умеренного состояния больного:
- Регулярный сбор анализов крови для контроля гемоглобина, эритроцитов и железа.
- Соблюдение рекомендованного образа жизни и питания.
- Понижение уровня железа в организме человека при помощи продуктов питания и лекарств.
При средней и большой форме талассемии разрабатывается ряд мероприятий по лечению:
- Постоянный контроль уровня гемоглобина и железа в составе крови при помощи анализов крови.
- Регулярное переливание крови для поддержания нужного количества эритроцитов в составе крови.
- Устранение лишнего железа из организма при помощи комплекса препаратов (Десферал).
- При большом увеличении селезенки рекомендовано ее резекция (удаление). Не применяется для детей младше пяти лет.
- Операция по пересадке костного мозга, которая на сегодня является лучшим способом лечения. Сложность этой процедуры заключается в индивидуальном подборе донора, подходящего по всем характеристикам.
- Ежедневный прием аскорбиновой кислоты для выведения железа из организма.
- Введение в рацион питания продуктов, снижающих всасывание железа в организм.
В чем опасность заболевания
- При малой форме этого заболевания больной подвержен минимальной опасности. Так как заболевание протекает практически бессимптомно и не имеет тяжелых последствий. Единственной опасностью является последующая передача аномального гена талассемии будущему поколению.
- При большой форме талассемии существует высокий процент смертности, особенно среди новорожденных больных. У больных зрелого и подросткового возраста существует опасность развития дополнительных тяжелых заболеваний, таких как рак, сепсис, инфекции.
Прогноз талассемии
Для больных талассемией прогноз зависит от формы тяжести заболевания:
- С малой формой талассемии люди часто проживают полноценную продолжительную жизнь. Отличие такого больного от здорового человека более слабым иммунитетом к различным инфекциям.
- Больные с тяжелой формой талассемии имеют обособленный образ жизни. У новорожденных с этим заболеванием все симптомы появляются на второй год жизни. Для людей с талассемией требуется регулярное переливание крови для поддержания функциональности организма. Часто последствиями талассемии является диабет, цирроз печени, инфаркт миокарда, тяжелые заболевания суставов.
- При появлении в семейном анамнезе этого заболевания возникает риск в дальнейшем распространении и заражении талассемии во всех последующих поколениях.
Профилактика
Основной целью профилактики является выявление фактора риска наследования талассемии.
Существует комплекс мероприятий, направленных на предупреждение и предотвращение этого заболевания:
- Дородовая диагностика и генетическая консультация, при которой выявляют предрасположенность родителей и будущего ребенка.
- Пренатальная диагностика. Проводится при обнаружении аномального гена у одного или обоих родителей. Иногда итогом такой диагностики может стать планированное прерывание беременности.
- Постоянное наблюдение больных с диагнозом талассемии у генетиков, гематологов и терапевтов.
- Больные талассемией обязаны постоянно соблюдать режим, помогающий избежать инфекций.